AG 6 Biochemistry and Molecular Medicine

Research Projects
Peroxisome biology and peroxisomal disorders
Many exciting research questions relating to biogenesis, function and pathophysiology of peroxisomes are still unanswered. Following up on our work on the intracellular transport of membrane proteins, the function of readthrough proteins, and on the role of peroxisomes in the heart and in neuronal tissue cells, we are focussing our interest on fhe function and biogenesis of peroxisomes, their role in pathophysiology, and on therapeutic approaches.
Peroxisomal disorders are caused by defects in peroxisome functions. Peroxisome biogenesis disorders (PBDs) include the Zellweger syndrome spectrum and rhizomelic chondrodysplasia punctata type 1. The Zellweger syndrome spectrum is a continuum of disorder characterized by developmental brain defects, skeletal and craniofacial dysmorphism, liver dysfunction, retinopathy, and sensorineural hearing loss.
Mutations in PEX10 are associated with a comparabl mild form of inheritable ataxia. We were given teh opportunity to contribute to the cellular charcterization of a PEX10 patient (Nava et al. 2022).
A new exciting field opened up, when we began studying peroxisomal calcium (Sargsyan et al 2021, Sargsyan et al. 2022).
We conducted the first super-resolution microscopy study of peroxisomes in wild-type human fibroblasts and cells derived from patients with a peroxisomal biogenesis disorder (Soliman et al. 2018).
Currently, we are studying peroxisome function in the heart, using stem cell and mouse models in the context of SFB1002.
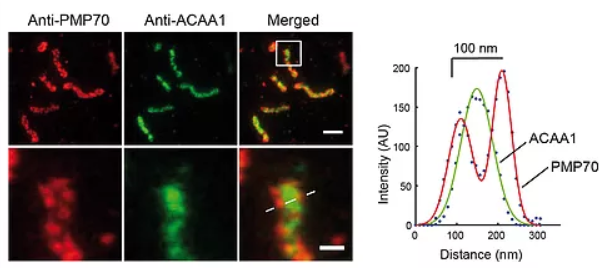
STED microscopy reveals the sub-diffraction morphology of peroxisomes. Two-color STED of peroxisome membrane and matrix. Immunofluorescence on human skin fibroblasts with anti-PMP70 (peroxisome membrane) and anti-ACAA1 (acetyl-CoA acyltransferase1, matrix). (Figure from Soliman et al. 2018).
Translational readthrough and readthrough therapy
FTR (functional translational readthrough) is a gene regulatory process in which stop codons are interpreted as sense codons during translation. By developing novel bioinformatics and screening techniques, we co-discovered the first human genes regulated by FTR: Lactate and malate dehydrogenase (LDH and MDH) are extended by several amino acids by FTR (Schueren et al. 2014; Hofhuis et al. 2016; Schueren and Thoms 2016]. The extensions (‘x’) contain functional targeting signals for peroxisomes.
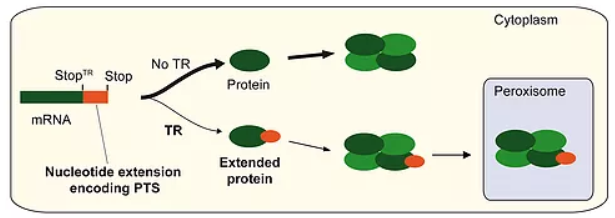
Endogenous translational readthrough – Exploring a new world of proteins. LDHBx, the extended form of LDHB, is generated by translational readthrough (TR) of the first stop codon during translation of the normal mRNA into protein. The interpretation of the stop codon as a sense codon leads to the prolongation of translation until the next in-frame stop codon is reached. This leads to the C-terminal addition of seven amino acids to the normal LDHB. The extension (between StopTR and Stop) harbors a peroxisomal targeting signal (PTS). Tetramers, which comprise the conventional subunits LDHB and/or LDHA together with a readthrough subunit LDHBx, can be transported into peroxisomes by the peroxisomal import machinery (piggyback import). The same mechanism combining readthrough and a hidden PTS1 after the first stop codon leads to the peroxisomal import of dimeric MDH1. The NCBI has adopted the definition of the x-nomenclature for readthrough proteins.
Based this FTR mechanism, novel peroxisomal redox shuttles may exist (lactate-pyruvate, and aspartate-malate shuttles). Readthrough-dependent metabolite shuttles can explain import of redox equivalents into the peroxisome and their export with mitochondria as the final destination.
The physiological readthrough is stimulated by a nucleotide consensus motif, which is also present in other genes that may be regulated by FTR.
LDHBx and MDH1x are only two of more than 50 new proteins that we have discovered to receive readthrough-extensions by a very leaky stop codon nucleotide context.
One goal of our research is to functionally and structurally understand the regulation of translational readthrough.
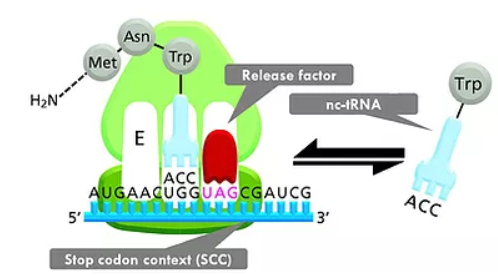
Translational readthrough. Competition of near-cognate tRNA (nc-tRNA) and release factor binding at the A-site of the ribosome is the basis of translational readthrough. In prokaryotes and eukaryotes, termination/release factors (eRF) mediate the termination of translation. By binding of nc-tRNA in place of eRF, the stop codon is read-through and an extended protein is formed. Normally, the equilibrium between release factor binding and (mis)incorporation of nc-tRNAs is >99.9% on the side of release factor binding and termination. Several conditions can shift this equilibrium: (1) We have identified stop codon contexts (SCCs), defined as stop codon with neighboring nucleotides that drastically shift the equilibrium towards nc-tRNA binding and continuation of translation so that the readthrough probability is increased by one or two orders of magnitude. This endogenous mechanism is universally conserved. (2) Aminoglycosides and other low molecular weight substances can bind to the small subunit of the ribosome and increase readthrough probability. This is a viable approach in the therapy of diseases caused by premature stop codon mutations.
We are also interested in characterizing the stop codon contexts (SCCs) in terms of how they contibute to the potential of translational readthrough-inducing drugs. With this approach, we aim to improve readthrough therapies of monogenetic diseases caused by premature termination codon mutations. As for vitrtually every mono genetic disorder, pathological stop mutations exist, a better understanding of the SCC-dependency of therapeutic success could avance the precision medizin of meny diseases. Not surprisingly, we hav started by analysing the pathologica SCCs of peroxisome biogenesis disorders (Schilff et al. 2021). We are currently including Rett syndrome stop codon mutations in our analysis.
Dysferlin and dysferlinopathies
Dysferlinopathies belong to the diverse group of muscular dystrophies and are caused by the absence of Dysferlin protein in skeletal muscle. The main two forms are Miyoshi myopathy (MM) and limb girdle muscular dystrophy type 2B (LGMD 2B). These diseases are characterized by progressive muscular weakness and wasting, typically starting in the second decade of life (Bulankina and Thoms, 2020). Interestingly, about half of the Dysferlin-deficient patients show an unusual disease phenotype by displaying increased levels of fitness before the onset of symptoms, which is not seen in any other form of muscular dystrophy. So far, no curative treatment is available.
The 230 kDa protein Dysferlin is composed of a C-terminal transmembrane domain and seven C2 domains. C2 domains are often associated with membrane binding and tubulating functions. Dysferlin has been shown to localize to the plasma membrane and the T-Tubule system in skeletal muscle and is known to act on intracellular membranes and to play a role in membrane repair during muscle regeneration (Bulankina and Thoms, 2020).
The T-tubule system is an extensive network formed by invaginations of the plasma membrane accounting for about 80% of the plasma membrane surface of striated muscle. Employing a wide variety of methods from in vitro reconstitution in liposomes, through cell models like human myoblasts, to mouse models, we could show that Dysferlin tubulates liposomes, generates a T-tubule-like membrane system in non-muscle cells, and links the recruitment of phosphatidylinositol 4,5-bisphosphate to the biogenesis of the T-tubule system (Hofhuis et al. 2017). Pathogenic mutants interfere with all of these functions, indicating that muscular wasting and dystrophy are caused by the Dysferlin mutants’ inability to form a functional T-tubule membrane system.
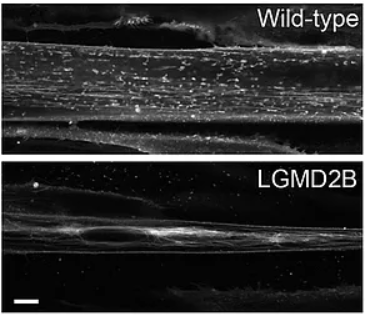
Dysferlin is required for T-tubule formation in vivo (and in vitro). The multi-C2 domain protein dysferlin localizes to the plasma membrane and the T-tubule system in skeletal muscle, however, its physiological mode of action is unknown. Mutations in the DYSF gene lead to autosomal recessive limb-girdle muscular dystrophy type 2B (LDMD2B). Tubule staining, using DiIC16(3), of human wild-type and dysferlin-deficient myoblasts, after differentiation in culture, shows compact tubular aggregates in dysferlin-deficient myoblasts that were not observed in control fibres. Scale bar: 10 µm. Figure from Hofhuis et al 2017.
Far less is known about the role and function of Dysferlin in the heart as dysferlinopathies are mainly considered as skeletal muscle diseases and only few patients suffer from cardiomyopathies. However, we showed that also in the heart, Dysferlin executes an important role in maintenance of the tubular system (TATS) (Hofhuis et al. 2020). By analysing Dysferlin expression and localization in healthy rat and mouse cardiomyocytes and Dysferlin-deficient mouse cardiomyocytes we were able to show that the protein localizes to the TATS in the heart and expression is upregulated during development. Absence of Dysferlin leads to massive structural reorganization and loss of transverse TATS structures resulting in defective calcium homeostasis and arrhythmia susceptibility that may have functional consequences in dysferlinopathy patients.